vispārinājums
Cistiskā fibroze ir visizplatītākā autosomālā recesīvā slimība Kaukāza populācijā, kas skar aptuveni 1 indivīdu katrā 2500.
Šis patoloģiskais stāvoklis ir zināms par kaitīgo ietekmi uz elpošanas sistēmu, bet tas ietekmē arī citas sistēmas, piemēram, gremošanas un reproduktīvās sistēmas.
Cilvēkiem ar cistisko fibrozi elpceļi ir aizsērējuši ar biezu, viskozu gļotu, ko ir grūti noņemt pat ar enerģiskāko klepu. Elpošana kļūst sarežģīta un pacienti - ja netiek veikti nepārtraukti centieni noturēt elpceļus vairākas reizes dienā - risks mirst viņu pašu sekrēcijas dēļ. Cistiskās fibrozes slimnieki bieži mirst pneimonijas dēļ, jo aizsērējusi elpceļi nodrošina auglīgu vidi baktēriju augšanai.

Cēloņi
Cistisko fibrozi izraisa cistiskās fibrozes fibrozes (CFTR) transmembrānas vadītspējas gēna mutācijas, kas lokalizētas 7. hromosomā (lokusa kartēšana: 7q31).
Ir zināmas vismaz 1500 mutācijas no CFTR gēna. Visbiežāk sastopamā mutācija parasti tiek saukta par "Delta-F508" (DF508), un to nosaka, izdzēšot 3 bāzes pārus eksonā 10, kas izraisa fenilalanīna zudumu pozīcijā 508.
Proteīna, ko kodē CFTR gēns, ir transmembrānas kanāls, kas pieder pie ATPāzes cilvēku tirdzniecības ģimenēm vai ABC transportieriem, kas atrodas epitēlija šūnu apikālās membrānas līmenī un tiek aizvietots ar hlora jonu transportēšanu.
Normālos apstākļos konkrētas šūnas, kas savieno elpceļus, izdalās gļotas kopā ar ūdens šķidrumu, kas samazina to blīvumu. Cistiskā fibrozē ūdens šķidruma sekrēcija tiek ievērojami samazināta , tāpēc gļotas kļūst ļoti blīvas un grūti noņemamas no elpošanas trakta.
Elpošanas epitēlijā, tāpat kā visi šķidrumus saturošie epitēliji, ūdens transportēšana ir atkarīga no šķīdinātāju transportēšanas. Lai izdalītu ūdeni, elpošanas epitēlija šūnas aktīvi transportē hlora jonus (Cl-) no intersticiālā šķidruma uz lūmenu, radot negatīvu elektrisko potenciālu, kas izraisa nātrija (Na +) pasīvo plūsmu tajā pašā virzienā. Na + un Cl kustības palielina šķidruma osmotisko spiedienu, kas mitrina epitēlija slīpumu, kas pagriezts pret lūmeni, līdz ar to ūdens pasīvi pārvietojas atbilstoši osmotiskajam gradientam, no intersticiālā šķidruma līdz lūmenim. Gēnu defekts, kas ietekmē cistiskās fibrozes rašanos, tieši kavē Cl- transportēšanu un netieši traucē Na + un ūdens transportēšanu. Līdz ar to epitēlijā neizveidojas ūdens sekrēcijai nepieciešamais osmotiskais gradients.
Riska faktori
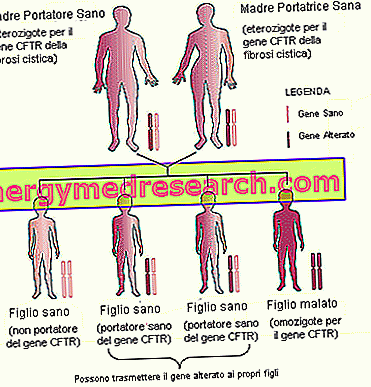
- Ģimenes mantojums . Ņemot vērā, ka cistiskā fibroze ir iedzimta slimība, kas tiek pārnesta autosomālā recesīvā veidā, ir svarīgi ņemt vērā topošo vecāku ģimenes anamnēzi.
- Šķirne . Cistiskās fibrozes biežums ir lielāks baltajiem cilvēkiem ziemeļos un Eiropā.

Simptomi un klīniskās pazīmes
Lai uzzinātu vairāk: Cistiskā fibrozes simptomi
Simptomu smagums var mainīties atkarībā no slimības gaitas: vairums klīnisko pazīmju ietekmē elpošanas sistēmu un kuņģa-zarnu trakta sistēmu.
Elpceļu pazīmes un simptomi
Ar cistisko fibrozi saistītā bieza un viskoza gļotāda izraisa elpceļu obstrukciju un kavē plaušu epitēlija līnijas blakusdobumu darbību, veicinot pašas gļotas izvadīšanu. Mazajos elpceļos radītā gļotādas stagnācija palīdz radīt ideālu vidi baktērijām, tādām kā Streptococcus aureus vai Pseudomonas aeruginosa, kas predisponē skartos pacientus ar atkārtotām infekcijām un iekaisumiem, kas savukārt var būt arī smags, kā pneimonija. Šo iekaisuma procesu hroniskais raksturs var kaitēt plaušu audiem laika gaitā un līdz ar to izraisīt sliktu elpošanas funkciju.
Tādējādi raksturīgie simptomi, kas attīstās cistiskās fibrozes gaitā, ietver:
- Noturīgs klepus ar krēpu ražošanu
- Sēkšana elpa
- Elpas trūkums un apgrūtināta elpošana
- Atkārtotas plaušu infekcijas
Gremošanas trakta pazīmes un simptomi
Biezās gļotas var arī aizsprostot kanālus, kas ved gremošanas fermentus no aizkuņģa dziedzera uz tievo zarnu. Bez šiem gremošanas fermentiem zarnas nevar sagremot un pilnībā absorbēt uztura uzturu (jo īpaši taukus, holesterīnu un taukos šķīstošos vitamīnus A, D, E un K).
Rezultāts bieži ir šāds:
- Pietūks vēders, meteorisms, steatoreja
- Slikts svara pieaugums un slikta izaugsme (nepietiekama uztura dēļ)
- Zarnu bloķēšana, īpaši jaundzimušajiem (meconium ileum)
- Aizcietējums vai smaga aizcietējums
Sarežģījumi
Elpošanas sistēmas komplikācijas
- Bronhektēzija : cistiskā fibroze ir viens no galvenajiem bronhektāzijas cēloņiem, tas ir stāvoklis, kas bojā elpceļus, izraisot obstruktīvu ventilācijas traucējumu, ievērojami samazinot izelpas plūsmu.
- Hroniskas infekcijas : bieza gļotas, kas stagnējas plaušās un deguna blakusdobumos, ir ideāla baktēriju un sēnīšu audzēšanas vieta. Tā rezultātā cilvēkiem ar cistisko fibrozi var būt biežas sinusīta, bronhīta vai pneimonijas uzbrukumi.
- Deguna polipi : tā kā deguna iekšējā odere ir iekaisusi un pietūkušas, var attīstīties mīkstas mīkstas augšanas (polipi), kas var kavēt elpošanu miega laikā.
- Klepus ar asinīm (hemoptīze): laika gaitā cistiskā fibroze var izraisīt elpceļu sieniņu retināšanu. Tā rezultātā pacienti ar cistisko fibrozi var izdalīt asinis no elpceļiem, parasti caur klepu.
- Pneimotorakss : šis stāvoklis, kas sastāv no gaisa uzkrāšanās pleiras dobumā, ir biežāk sastopams gados vecākiem cilvēkiem ar cistisko fibrozi. Pneumotorakss var izraisīt sāpes krūtīs un elpas trūkumu.
- Plaušu sabrukums : hroniskas plaušu infekcijas var bojāt plaušu parenhīmu, padarot plaušu sabrukumu visticamāk.
- Elpošanas mazspēja : laika gaitā cistiskā fibroze var bojāt plaušu audus un apdraud tās funkcionalitāti.
Gremošanas sistēmas komplikācijas
- Uztura trūkumi : biezas gļotas var bloķēt aizkuņģa dziedzera kanālus, kas satur gremošanas fermentus no aizkuņģa dziedzera uz tievo zarnu. Bez šiem fermentiem organisms nespēj absorbēt proteīnus, taukus un taukos šķīstošos vitamīnus.
- Diabēts : aizkuņģa dziedzera beta šūnas izdalās insulīnu, kas ir nepieciešams, lai regulētu glikozes līmeni asinīs. Cistiskā fibroze palielina diabēta attīstības risku. Apmēram 20% no skartajiem cilvēkiem diabēta ārstēšanā ir sasnieguši 30 gadu vecumu, tāpēc pacientiem regulāri pārbauda glikozes līmeni asinīs.
- Žultsvadu traucēšana : kanāls, kas nes žults no aknām un no žultspūšļa uz tievo zarnu, var būt bloķēts un iekaisis, izraisot aknu darbības traucējumus un dažreiz žultsakmeņus.
- Taisnās zarnas prolapss : bieža klepus vai slodze izkārnījumos (aizcietējums) var izraisīt iekšējo taisnās zarnas audu izplūdi no tūpļa.
- Intussusception : bērniem ar cistisko fibrozi ir augstāks intussuscepcijas risks, stāvoklis, kurā daļa zarnu krokām atlocās uz sevi, radot invaginācijas. Rezultāts ir zarnu obstrukcija, ārkārtas situācija, kas prasa tūlītēju ārstēšanu.
Reproduktīvās sistēmas komplikācijas
Gandrīz visi vīrieši ar cistisko fibrozi ir sterili, jo cauruļvadi, kas savieno sēkliniekus un prostatas dziedzeri (vas deferens), ir bloķēti ar gļotām vai vispār trūkst (cauruļvadu trūkums joprojām ir nezināms iemesls).
Lai gan sievietes ar cistisko fibrozi var būt mazāk auglīgas nekā citas sievietes, tās joprojām saglabā spēju iedomāties un pārtraukt grūtniecību. Tomēr grūtniecība var veicināt cistisko fibrozes simptomu pasliktināšanos.
Citas komplikācijas
Osteoporoze : cilvēkiem ar cistisko fibrozi ir lielāks osteoporozes attīstības risks, līdz ar to zaudējot masu un kaulu stiprumu. Pacientiem jāiesaka lietot atbilstošu kalciju, D vitamīnu un bisfosfonātus.
Elektrolītu nelīdzsvarotība : cilvēkiem ar cistisko fibrozi sviedriem parasti ir augstāks sāls līmenis. Pazīmes un simptomi, piemēram, sirdsdarbības ātruma palielināšanās, nogurums, vājums un zems asinsspiediens, palīdz uzsvērt dekompensāciju, kas notiek ar sālsūdens sāls līdzsvaru asinīs.
Cistiskās fibrozes diagnostika un terapija »


