Kas ir Marfana sindroms?
Marfana sindroms apraksta sarežģītu saistaudu slimību, kas galvenokārt skar acis, sirds un asinsvadu sistēmu un skeleta muskuļu sistēmu. Tomēr, ņemot vērā, ka katrs orgāns sastāv no saistaudiem, Marfana sindroms var ideāli iznīcināt un traucēt katras anatomiskās vietas augšanu un darbību.
Šis sindroms tiek pārnests kā autosomāla dominējošā iezīme: tāpēc mēs saskaramies ar nopietnu ģenētisku slimību, kam ir ļoti mainīga fenotipiskā izpausme (defekti var ļoti atšķirties no ģimenes uz ģimeni vai no pacienta uz pacientu).
Kas izraisa Marfana sindromu, ir FBN1 gēna pārveidošana (15. hromosomā), kas kodē fibrilīnu-1, kas ir ļoti svarīgs saista glikoproteīns, kas veido strukturālu atbalstu mikrofibriliem.
Mikrofibrili: kas sastāv no fibrilīna, mikrofibrīli atrodas ekstracelulārajā matricā, kurā tie veido audumu elastīna nogulsnēšanai elastīgajās šķiedrās. Lai gan mikropibrīli ir visuresošs ķermenis, tās ir lielākoties cirvju ķermeņu aortas, saites un zonulas (acu līmenī).
Marfana sindroms ir autosomāla dominējošā slimība, tikai bērni, kuri ir pārgājuši FBN-1 gēnu, ko mainījuši abi vecāki. Tomēr vienā gadījumā no 4 slimība ir spontānu mutāciju rezultāts pacientiem, kuriem nav ģimenes anamnēzes.
Slimības nosaukums nāk no Francijas pediatra, kurš to pirmo reizi aprakstīja 1896. gadā (A. Marfan), pēc tam tai bija jāgaida līdz 1991. gadam, lai identificētu izmainīto gēnu, kas iesaistīts simptomātiskajā izpausmē: atklājējs bija F. Ramirez.
Skatieties video
X Skatiet videoklipu vietnē YouTubeCēloņi
Mēs esam minējuši, ka Marfana sindroms ir tūlītēja gēna mutācijas izpausme, kas kodē fibrilīnu-1. Mēģināsim padziļināt tēmu, lai saprastu, kurš mehānisms ir izveidots sindroma izraisīšanai.
FIBRILLINA 1 ir elastīna glikoproteīna komponents, kas ir būtisks elastības un audu izturības nodrošināšanai un saglabāšanai. Fizioloģiskos apstākļos fibrilīns 1 saistās ar citu proteīnu, kas pazīstams kā TGF-beta (vai transformējošais augšanas beta faktors). Šķiet, ka TGF-beta ir iesaistīts kaitīgos procesos, kas ietekmē asinsvadu gludo muskuli un ekstracelulāro matricu. Sākot no šiem pieņēmumiem, daži autori ir pārliecināti, ka Marfana sindroms papildus FBN-1 gēna mutācijai ir saistīts arī ar TGF-beta pārpalikumu, īpaši aortā, sirds vārstuļos un plaušās.
biežums
Tiek lēsts, ka Marfana sindroms skar 1 subjekti katru 3000-5000 dzimušo un izpaužas kā neskaidrs starp vīriešiem un sievietēm, nenosakot precīzu šķirni. Statistika liecina, ka 75% pacientu ir pozitīva ģimenes vēsture; atlikušajos 25% cēlonis ir sporādiskas mutācijas, kas, šķiet, kaut kādā veidā ir saistītas ar tēva vecumu koncepcijas laikā.
Bērniem ar ārkārtīgi smagām Marfana sindroma formām paredzamais mūža ilgums ir mazāks par vienu gadu.
Pirms atvērto sirds ķirurģisko stratēģiju attīstības, vairumam pacientu ar Marfana sindromu vidējais dzīves ilgums bija 32 gadi; pateicoties pastāvīgai medicīnisko un farmakoloģisko terapiju uzlabošanai, pašlaik Marfana sindroma pacienti dzīvo vidēji līdz 60 gadiem.
Pazīmes un simptomi
Lai uzzinātu vairāk: Simptomi Marfana sindroms
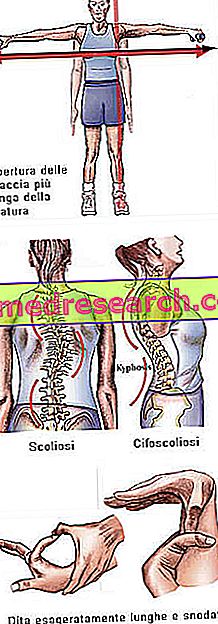
Marfana sindroms var būt pilnīgi asimptomātisks. Ietekmētajiem pacientiem ir pārlieku tieva struktūra, kas ir nesamērīgi augsta un plāna. Apakšējo un augšējo ekstremitāšu garums ir daudz lielāks nekā stumbrs (dolicostenomegalia). Ir arī runāts par arachnodactyly, lai vislabāk izteiktu pārspīlēto pirkstu garumu, kas raksturīgs Marfana sindroma skartajiem cilvēkiem, tāpēc rokas salīdzina ar zirnekļa kājām.
Attiecībā uz augstumu šiem pacientiem ir vidējais augstums virs 97. procentiles.
Citas raksturīgas pazīmes, kas bieži sastopamas pacientiem ar Marfana sindromu, ir:
- Ieroči atveras augstāk par augstumu
- Loose locītavas → pārspīlēta locītavu mobilitāte
- Krūškurvja sienas deformācija
- Kristāliskā pārvietošana
- Augšējā ķermeņa daļa ir mazāk attīstīta nekā apakšējā daļa
- Spontāns pneimotorakss (11%)
- skolioze
- Ādas strijas pie augšstilba, muguras, deltveida, krūšu līmeņa
Starp visbiežāk sastopamajām pazīmēm, kas saistītas ar Marfana sindromu, pieminējām sirds vārstuļa prolapsu un mitrālo vārstu nepietiekamību: līdzīgs stāvoklis var viegli veicināt aortas gredzena paplašināšanos un aortas dalīšanu.
Tabulā redzamas pazīmes, kas var rasties pacientiem ar Marfana sindromu. Tur aprakstītās rakstzīmes ne vienmēr ir klāt, bet labas no tām var atrast.
| Ietekmētā anatomiskā vieta | Iespējamie simptomi |
gudrs | Kāpēc krūškurvja, jostas un sakrālās zonā |
Acis | Redzes traucējumi, astigmatisms, tīklenes atdalīšanās, šaura leņķa glaukoma, lēcas dislokācija, tuvredzība |
Kaulu struktūra | Artralģija, kyphoscoliosis, dolicostenomelia (ekstremitātes attīstība ekstremitāšu garumā attiecībā pret stumbru), hipermobilitāte, augsta aukslējas, deformētas krūtis, plakanas kājas, šauras un plānas plaukstas, patoloģiska atgriešanās / krūšu kaula izvirzījums, skolioze, izliekti pleci, spondylolisthesis |
Dita | arachnodactyly |
plaušas | Spontāna pneimotorakss, aizdusa, idiopātiska plaušu obstruktīva slimība |
Sejas izmaiņas | Ogvas aukslējas (aukslēju malformācija), mandibulārā retrognathia (žokļa attīstības defekts), iegarena seja |
sirds | Stenokardija, vēdera aortas aneirisma, sirds aritmija, krūšu aorta dilatācija / plīsums / dissekcija, aortas nepietiekamība, mitrālā vārsta prolapss |
valoda | Valodas grūtības |
diagnoze
Ņemot vērā vairāk nekā 200 iespējamās mutācijas, ģenētisko marķieru izmantošana diagnostikas nolūkos ir gandrīz neiespējama.
Marfana sindroma noteikšana ne vienmēr ir tik tūlītēja, ņemot vērā to, ka mutācijas fenotipiskā izpausme ne vienmēr ir acīmredzama un vienkārši identificējama. Diagnostiskā aizkavēšanās var nopietni apdraudēt pacienta izdzīvošanu: vienkārši domājiet, piemēram, par nespēju atpazīt sirds un asinsvadu problēmu.
Marfana sindroma diagnostikas kritēriji tika izstrādāti starptautiski 1996. gadā: diagnoze ir ģimenes anamnēzes izpēte, kas saistīta ar sindroma nozīmīgu un nelielu rādītāju kombināciju.
Daži no izmantotajiem diagnostikas testiem ir šādi:
- ehokardiogrāfija
- Magnētiskā angiorisonance un CT (aortas izmeklēšanai)
- magnētiskās rezonanses angiogrāfija (MRA) ar kontrasta šķidrumu (lai izveidotu aorta iekšējās struktūras)
- pārbaude ar spraugas lampām (lai analizētu objektīva iespējamo novirzi)
- acu spiediena mērīšana (lai parādītu iespējamo glaukomas klātbūtni)
- ģenētiskie testi (ieteicams pirms bērna ierašanās, lai noskaidrotu sindromu vai nē)
terapiju
Tā kā tā ir ģenētiska slimība, nav nevienas zāles vai ārstēšanas, kas var izraisīt slimības nomaiņu.
Tomēr narkotiku lietošana ir būtiska, lai mazinātu simptomus un izvairītos no komplikācijām, jo īpaši sirdsdarbības. Šim nolūkam īpaši piemēroti ir medikamenti asinsspiediena samazināšanai, piemēram, sartāni (īpaši), AKE inhibitori un beta blokatori.
Marfana sindroma kontekstā pacienti, kas arī cieš no skoliozes, var sekot īpašai aprūpei, kā arī tiem, kurus skārusi glaukoma.
Ķirurģija ir iespējama, lai izlabotu anomālu aortas dilatāciju, kas bieži vien apvieno lielāko daļu pacientu ar Marfana sindromu.
Turpināt: Marfana sindroms - narkotikas un kopšana »